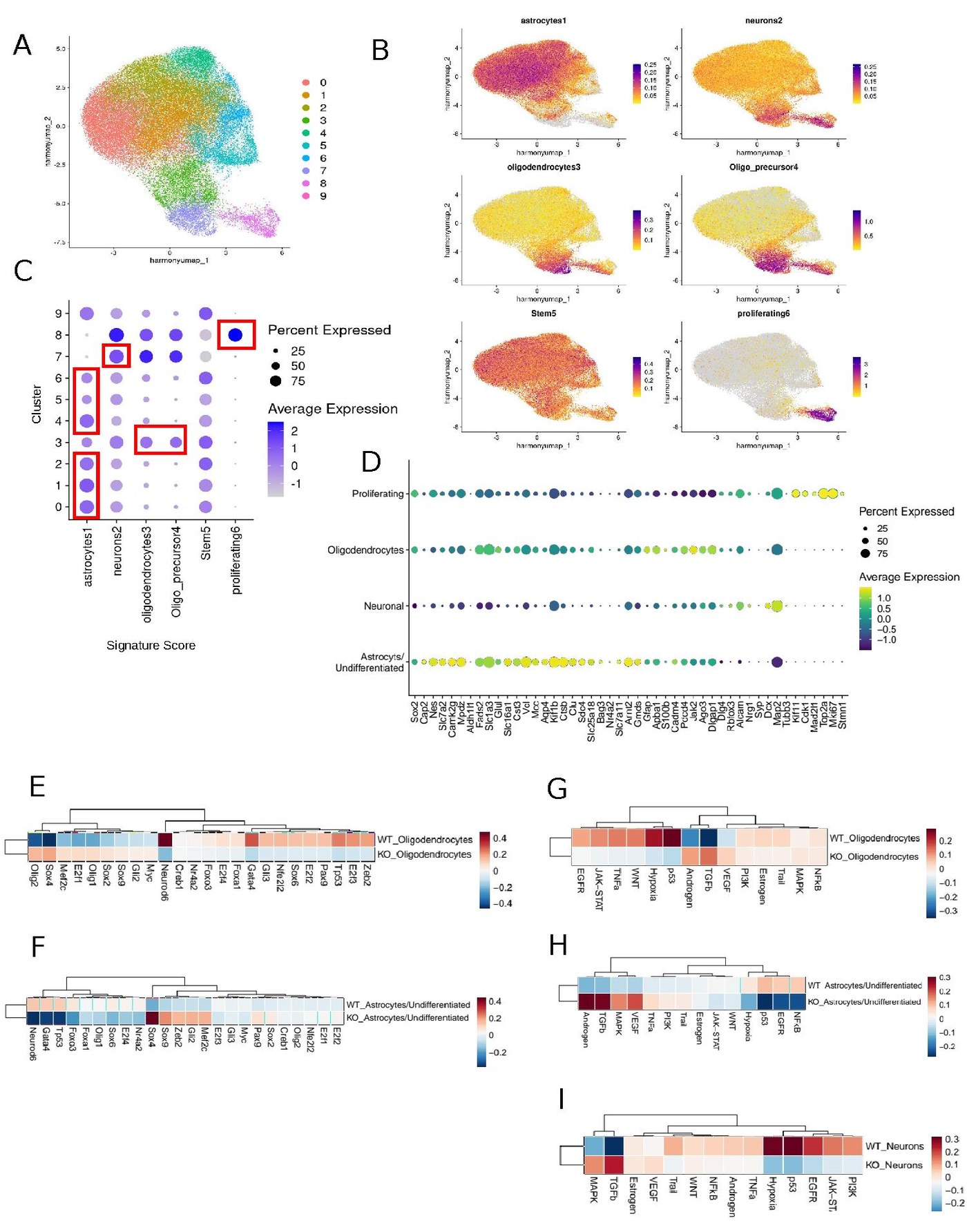
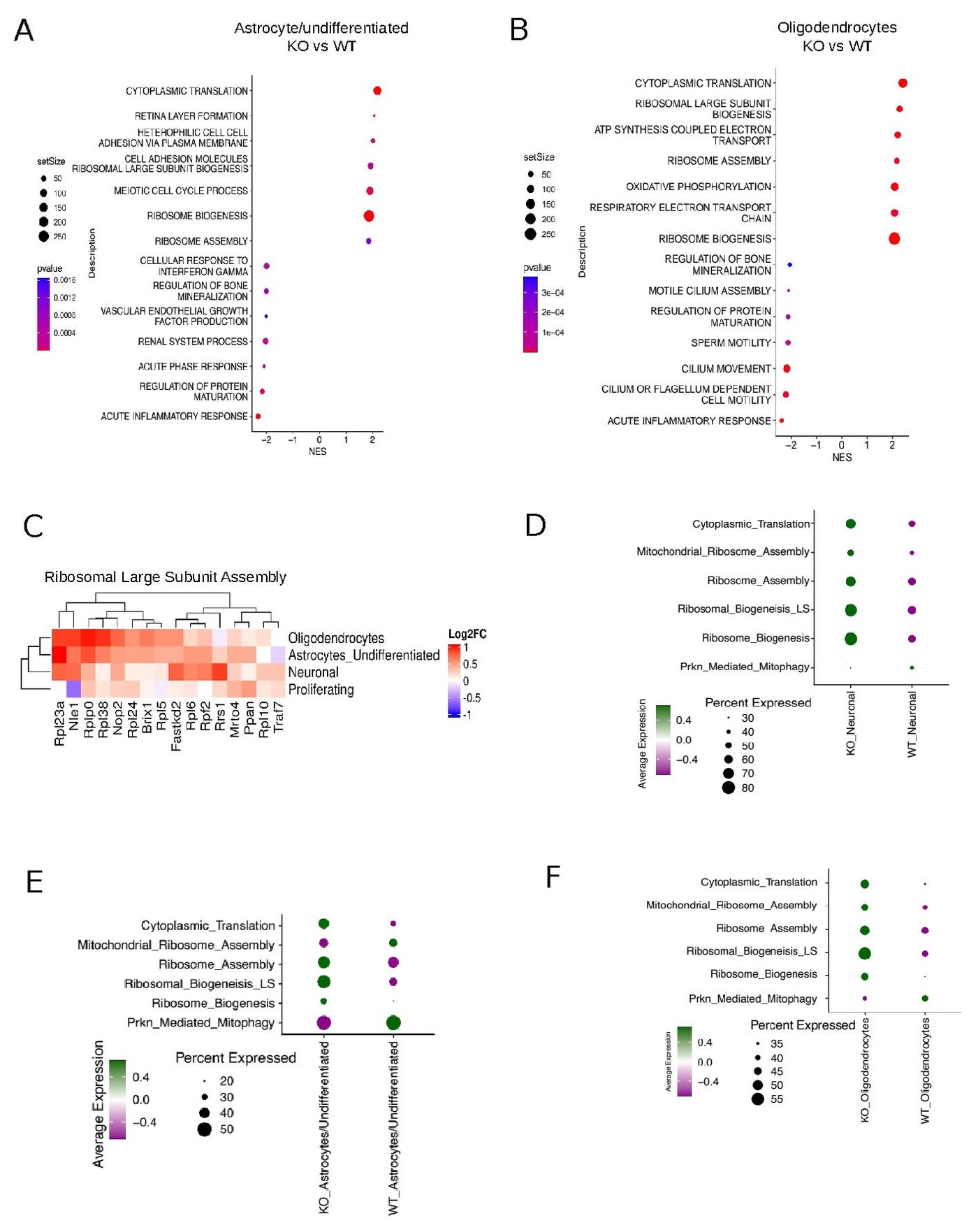
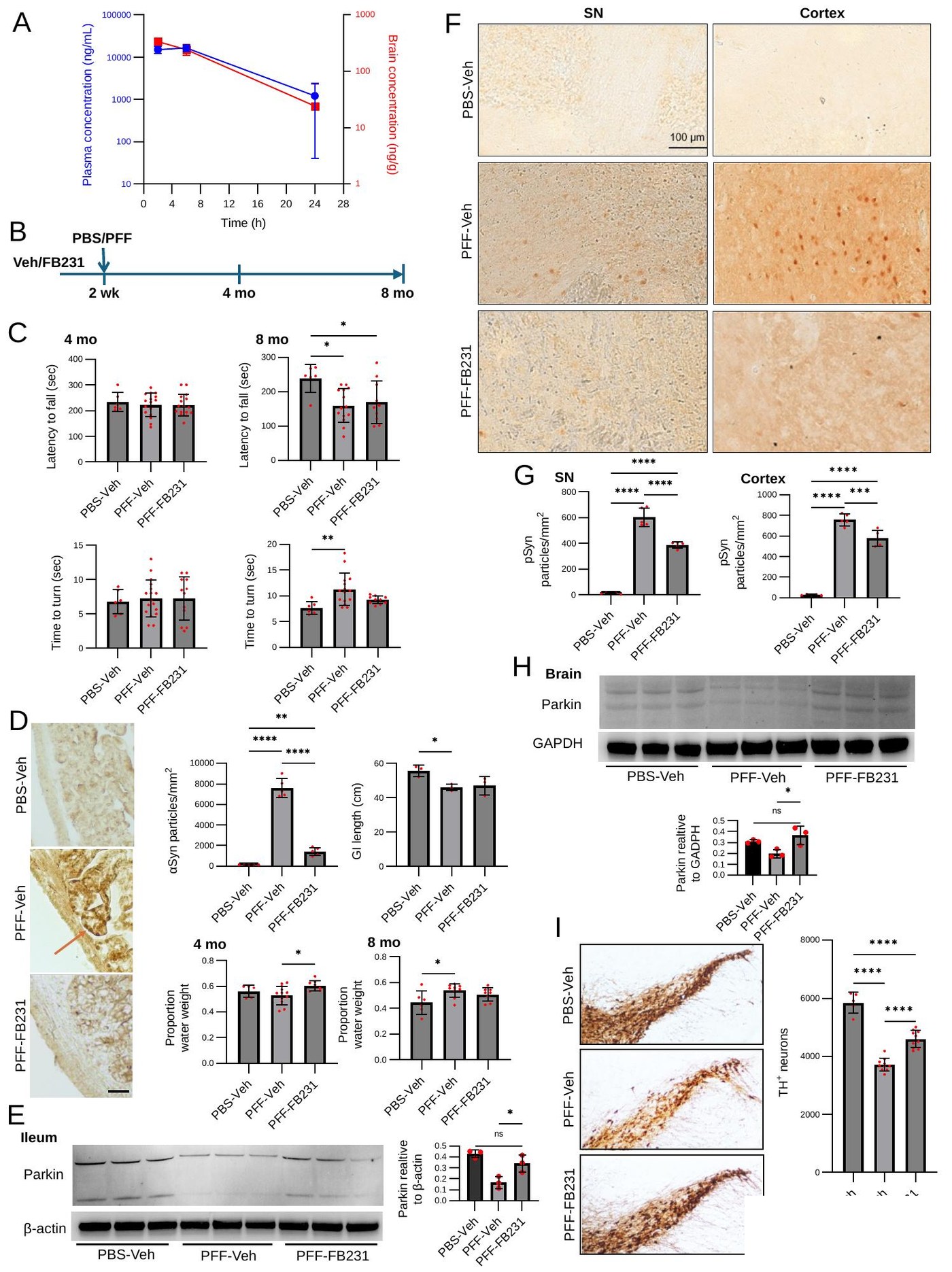
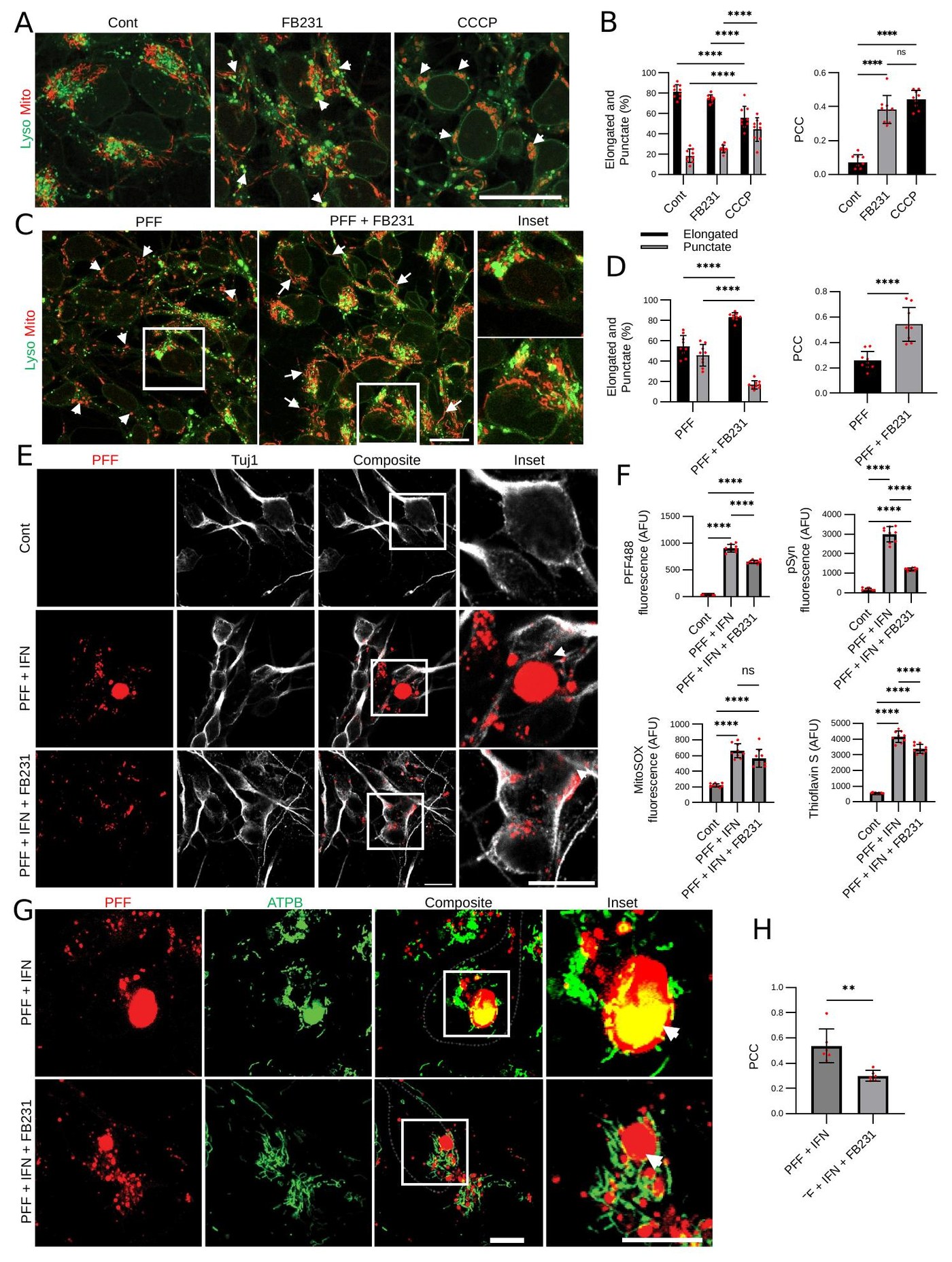
From Sample to Biology — A Reproducible NGS Workflow
For every iPSC-based study, I run a standardized pipeline: total-RNA extraction (Qiagen RNeasy or Trizol — QC by Bioanalyzer RIN ≥ 8), library prep (Illumina Stranded mRNA or 10x Chromium v3), sequencing (NovaSeq 6000 or NextSeq 2000), and analysis with STAR/Salmon → DESeq2 / edgeR (bulk) or Cell Ranger → Seurat / Scanpy (scRNA-seq). Pathway analysis layers on GSEA (fgsea, MSigDB Hallmark/KEGG/Reactome) and transcription-factor activity inference with DecoupleR. All findings are cross-validated by qPCR, Western, and imaging.
For bulk: Qiagen RNeasy Mini with on-column DNase or Trizol phase separation. For single-cell: dissociate to single-cell suspension with viability >85%. QC by Bioanalyzer/TapeStation; reject samples with RIN < 7.
Bulk: Illumina Stranded mRNA with poly-A selection or NEBNext rRNA-depleted. Single-cell: 10x Chromium Single Cell 3' v3.1. Validate library size on TapeStation; quantify by Qubit/qPCR before pooling.
Bulk: PE150, ~30 M reads/sample minimum (more for low-expressed genes). Single-cell: PE150 with 10x indexing, ~20–50k reads/cell.
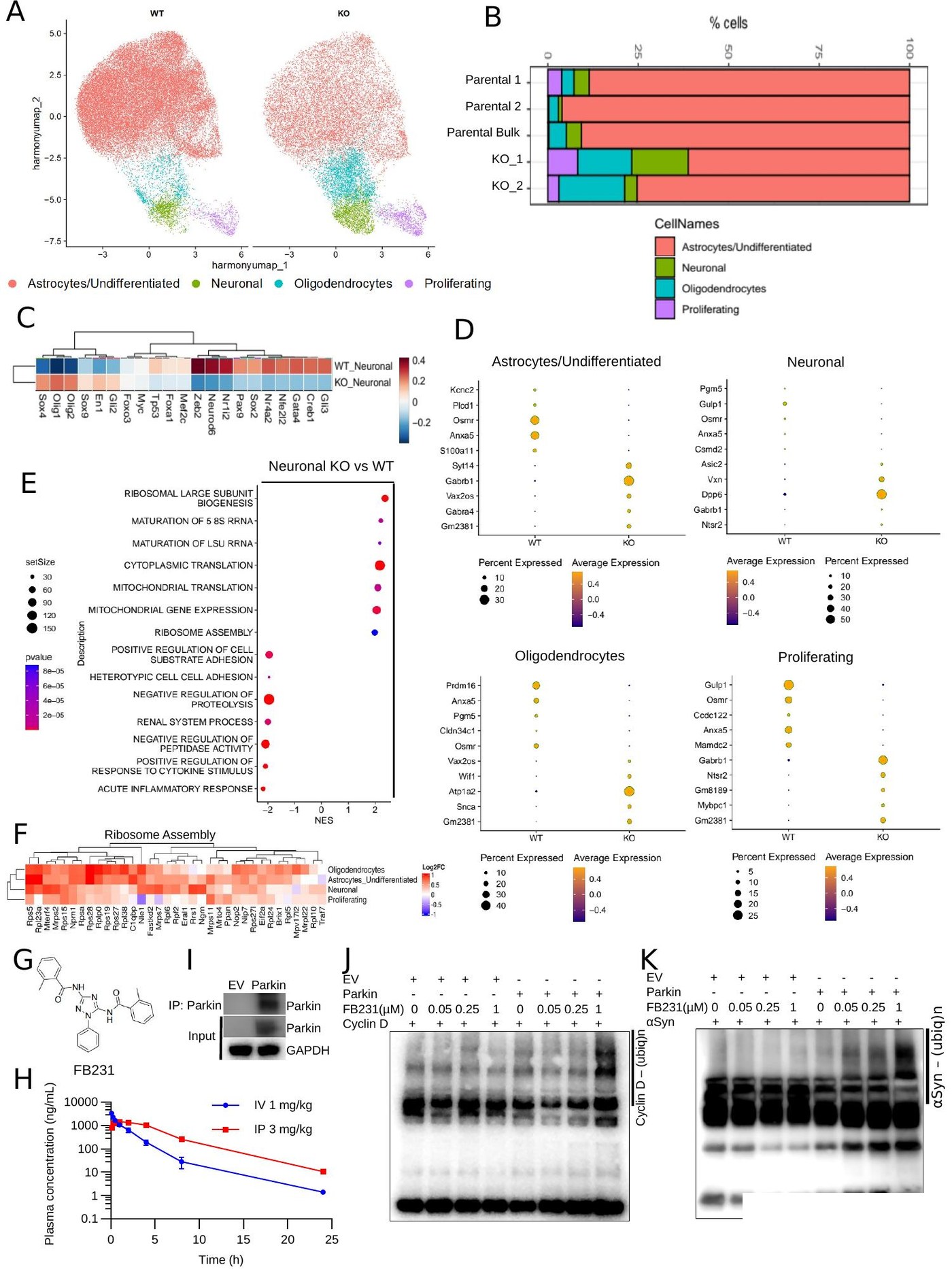
Bulk: STAR against GRCh38 or Salmon for rapid quasi-mapping; quantify counts → DESeq2 / edgeR. Single-cell: Cell Ranger → Seurat v5 or Scanpy + Harmony batch correction.
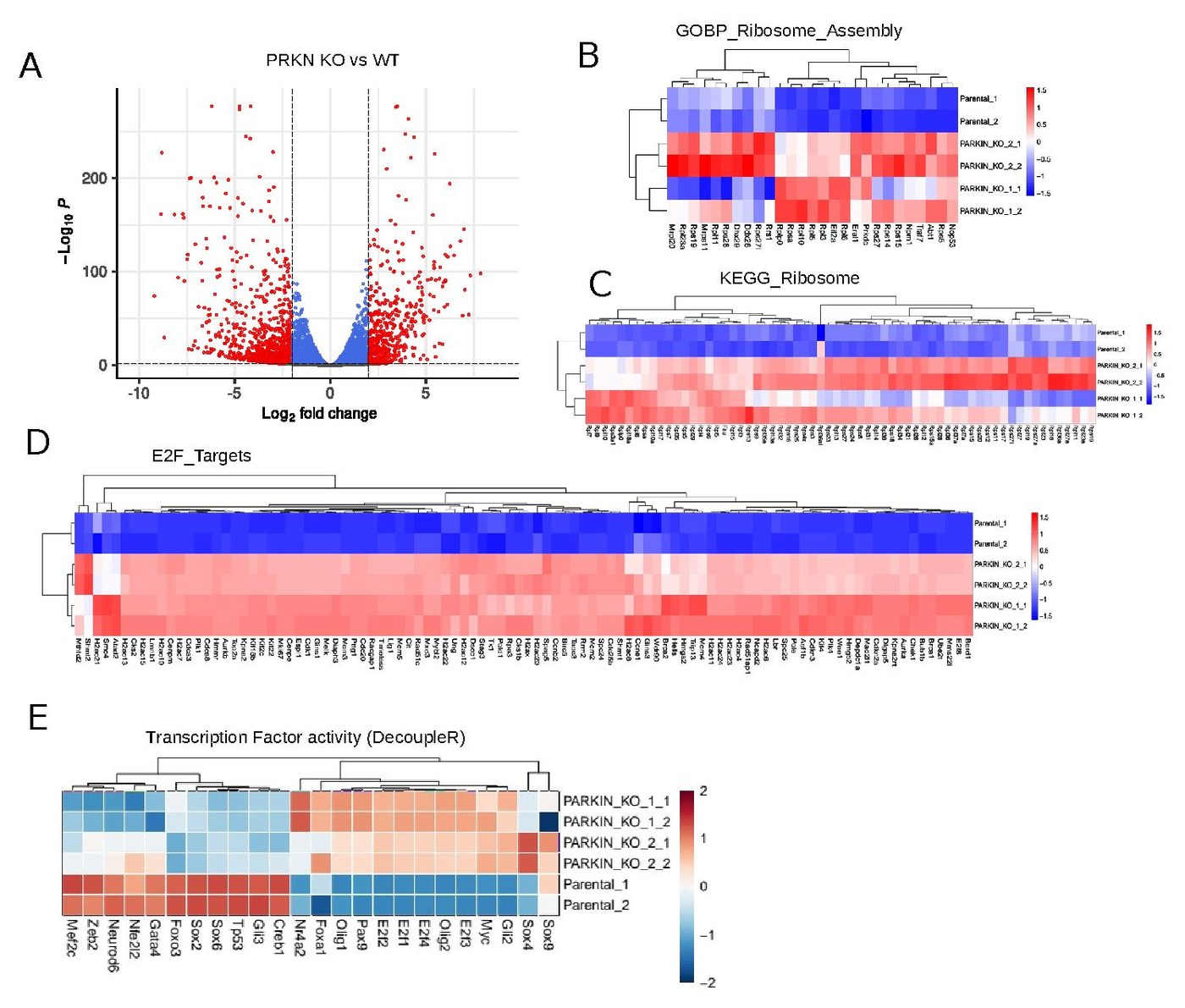
fgsea against MSigDB Hallmark, KEGG, GOBP, Reactome with 1k–10k permutations (NES > |1.5|, FDR < 0.05). DecoupleR with CollecTRI / DoRothEA regulons for TF activity.